
Abstract
In contemporary clinical oncology, radiolabeled octreotide analogs have demonstrated remarkable efficacy in cancer imaging and therapy. However, the broader application of radiopeptide-based paradigms is significantly limited by the inherent metabolic instability of systemically administered peptide analogs. This study explores the hypothesis that the concurrent in vivo administration of specific enzyme inhibitors can enhance peptide bioavailability, thereby improving tumor uptake. Through the co-injection of phosphoramidon (PA), a neutral endopeptidase inhibitor, we observed substantial increases in the circulating levels of intact somatostatin, gastrin, and bombesin radiopeptides in murine models. This protection strategy resulted in a notable augmentation of radiopeptide uptake in tumor xenografts within mice.
Methods: Peptide conjugates including [DOTA-Ala1]SS14, PanSB1, and DOTA-MG11 were labeled with 111In using a 20-minute heating process at an acidic pH. Metabolic stability was assessed via high-performance liquid chromatography (HPLC) analysis of blood samples obtained 5 minutes post-injection of the radiopeptide, both with and without PA co-administration. Biodistribution studies were conducted in tumor-bearing severe combined immunodeficient (SCID) mice, evaluating the uptake of each 111In-labeled radiopeptide alone and in conjunction with PA.
Results: The presence of PA significantly improved the metabolic stability of [111In-DOTA-Ala1]SS14, elevating the percentage of intact radiopeptide in circulation after 5 minutes from less than 2% to 86%. This enhanced protection translated to a substantial increase in AR4-2J xenograft uptake, rising from under 1 percentage injected dose per gram of tissue (%ID/g) to 14 %ID/g at 4 hours post-injection. Similarly, PA co-administration markedly increased circulating intact 111In-PanSB1 from 12% to 80% at 5 minutes, and tumor uptake in human PC-3 xenografts escalated from less than 4 %ID/g to over 21 %ID/g at 4 hours. Consistent results were observed with [111In-DOTA]MG11, where PA co-administration boosted intact peptide levels from below 5% to 70% at 5 minutes, leading to a significant uptake increase from 2 %ID/g to over 15 %ID/g in both AR4-2J and A431(CCKR+) tumor models at 4 hours. SPECT/CT imaging corroborated these findings, clearly visualizing enhanced tumor targeting in AR4-2J tumor-bearing mice at 4 hours when PA was co-administered.
Conclusion: This study unequivocally demonstrates that the co-administration of enzyme inhibitors effectively extends the in vivo survival of radiolabeled peptides, ensuring their protected transit to the target site. This “serve to protect” approach significantly amplified radiotracer accumulation in tumor xenografts, potentially leading to improved diagnostic sensitivity and therapeutic efficacy of radiopeptide drugs in cancer patients. These findings unveil promising avenues for utilizing biodegradable (radio)peptide drugs of both natural and synthetic origins and for designing metabolically stable analogs.
The selective overexpression of peptide receptors on cancer cell membranes, compared to their lower expression in adjacent healthy tissues, provides a unique opportunity for targeted cancer visualization and treatment using radiolabeled peptide analogs, known as radiopeptides (1–3). These radiopeptide drugs are meticulously engineered to deliver radionuclides specifically to their receptor targets at primary and metastatic tumor sites. Diagnostic radionuclides, emitting gamma photons detectable by SPECT or PET cameras, facilitate highly specific tumor lesion visualization. Therapeutic radionuclides, emitting particle radiation, can induce apoptosis and cell death in tumor cells (4–6). The clinical success of [111In-diethylenetriaminepentaacetic acid (DTPA)]octreotide in diagnosing somatostatin receptor-positive neuroendocrine tumors (7) and the Phase 3 clinical trials of [177Lu-DOTA0,Tyr3]octreotate for somatostatin receptor-targeted therapy (8,9) underscore the potential of this approach. Furthermore, numerous other regulatory peptide receptors, abundantly expressed in various cancer types, are emerging as promising targets for novel radiopeptide ligands (3).
The effectiveness of radiopeptide therapy hinges on the secure delivery of intact radiopeptides to their receptor targets following intravenous administration. Upon entering the bloodstream, radiopeptide integrity is immediately challenged by ubiquitous proteolytic enzymes present in blood or anchored to blood cells and vascular walls. Transit through major organs such as the liver, lungs, kidneys, and gastrointestinal tract further exposes radiopeptides to a higher concentration of degrading enzymes. The intricate interplay between native peptide ligands, their receptors, and endogenous enzymes is well-established in both normal physiology and cancer biology. Traditional strategies to enhance radiopeptide stability have focused on structural modifications, including amino acid substitutions, bond modifications, cyclization, and multimerization (10,11). However, these approaches are often resource-intensive, time-consuming, and may result in analogs with suboptimal pharmacological properties.
We propose an alternative, unexplored strategy: employing enzyme inhibitors to optimize radiopeptide delivery to tumor-associated receptors, aiming to enhance diagnostic sensitivity and therapeutic efficacy against both primary and metastatic cancer. By co-injecting a single inhibitor of neutral endopeptidase (NEP; EC 3.4.24.11; neprilysin; CD10) (12) in animal models, we achieved significant increases in the circulating levels of intact radiopeptide analogs from the somatostatin, gastrin, and bombesin families. Crucially, this strategy facilitated, for the first time, a notable amplification of radiopeptide uptake in diverse tumor xenografts expressing the corresponding receptor targets. This “serve to protect” mechanism ensures the radioligands reach their intended destination with greater efficacy.
Specifically, we intravenously administered phosphoramidon (PA) alongside different radiopeptides in mice. PA is a potent, reversible, and competitive NEP inhibitor, with its binding structure to human NEP well-characterized (13). Its high water solubility allows for bolus injection with radiopeptides, ensuring effective in vivo NEP inhibition. Originally isolated from Streptomyces tanashiensis cultures, convenient synthetic methods for PA have recently become available (14,15).
This study included three 111In-labeled, DOTA-modified radioligands representing somatostatin-14, bombesin, and gastrin peptide types. First, we investigated PA co-injection effects on the stability and localization of [111In-DOTA-Ala1]SS14 in somatostatin subtype 2 receptor (sst2)-expressing AR4-2J tumors in mice (16). Second, we examined the bombesin analog 111In-PanSB1 ([111In-DOTA-PEG2-dTyr-Gln-Trp-Ala-Val-βAla-His-Phe-Nle-NH2), designed based on a previously reported pan-bombesin receptor binding sequence (17–19). PA’s impact on in vivo stability and tumor uptake was evaluated in human PC-3 xenografts expressing the gastrin-releasing peptide receptor (GRPR) in mice (20,21). Third, we explored the gastrin-cholecystokinin peptide family using [111In-DOTA]MG11 (111In-DOTA-dGlu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2). We assessed PA co-injection effects on its in vivo stability and uptake in two tumor models: A431-CCK2R(+) and A431-CCK2R(−) xenografts (generated from A431 cells transfected to express human cholecystokinin subtype 2 receptor [CCK2R] and control A431 cells without CCK2R expression, respectively), and the CCK2R-expressing AR4-2J tumor model (22–24). [111In-DOTA]MG11 was previously reported to exhibit low renal accumulation but lower metabolic stability and tumor targeting compared to full-chain gastrin analogs (23,25).
Our findings highlight NEP’s significant role in the in vivo processing of N-terminal radiometallated peptides, consistent with its widespread presence in the human body (12). The importance of NEP in the catabolism of radiopeptides tested, and potentially other radiopeptide ligand classes, warrants further investigation. This preliminary study aims to stimulate further research into the critical roles of major peptidases in the breakdown of intravenously administered (radio)peptide drugs.
MATERIALS AND METHODS
Materials and Instrumentation
All chemicals were reagent grade. Peptides were synthesized or commercially sourced. [DOTA-Ala1]SS14, [Tyr3]octreotate (H-dPhe-c[Cys-Tyr-dTrp-Lys-Thr-Cys]-Thr-OH), and demogastrin 2 [N4-Gly-dGlu-(Glu)5-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2] were synthesized via previously described solid-phase methods (16,24). PanSB1 and DOTA-MG11 were acquired from PiChem, and [Tyr4]bombesin (Pyr-Gln-Arg-Tyr-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2) from Bachem AG. PA [*N*-(α-rhamnopyranosyloxyhydroxyphosphinyl)-l-leucyl-l-tryptophan × 2Na × 2H2O] was purchased from PeptaNova GmbH. 111InCl3 in 50 mM HCl (370 MBq/mL on calibration date) was obtained from Mallinckrodt Medical BV for 111In labeling.
Radiochemical HPLC analyses were performed using a Waters chromatograph with a model 600 solvent delivery system, coupled with a Waters model 996 photodiode-array UV detector and a Raytest Gabi γ detector. Waters Millennium Software controlled the HPLC system and processed data. Radioactivity was measured using a Canberra Packard Auto-Gamma 5000 series automatic well-type γ counter (NaI(Tl) 3′′-crystal). SPECT/CT images were acquired using a 4-head multipinhole NanoSPECT/CT camera (Bioscan Inc.) at the Erasmus MC Animal Imaging Facility.
111In Labeling and Quality Control
111In labeling was performed by adding 10 nmol of peptide analog to 37–74 MBq of 111InCl3 in 0.1 M sodium acetate buffer and 10 mM sodium ascorbate, achieving a typical end pH of 4.6. Reactions were heated in a boiling water bath for 20 minutes for complete labeling (26). Before HPLC quality control, 1 mM ethylenediaminetetraacetic acid in 0.1 M acetate buffer was added to scavenge free 111In3+ (27). For DOTA-MG11, 5 μL of 0.1 M methionine solution was added to prevent Met residue oxidation (28).
Radiochemical analyses used an XBridge Shield RP18 column (5 μm; 4.6 × 150 mm; Waters) with a matching guard column. Elution was performed at 1 mL/min using a linear gradient of 0.1% trifluoroacetic acid–water and acetonitrile (10% acetonitrile initially, increasing 1% per minute).
Cell Cultures
The rat pancreatic tumor cell line AR4-2J, expressing sst2 and rat gastrin/CCK2R, was kindly provided by Stephen Mather (St. Bartholomew’s Hospital, London, U.K.) and cultured as previously described (29,30). Human androgen-independent prostate adenocarcinoma PC-3 cells (LGC Promochem), expressing human GRPR (20), were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with GlutaMAX-I (Gibco BRL Life Technologies), supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Human epidermoid carcinoma cell lines A431-CCK2R(+) and A431-CCK2R(−), gifted by Otto C. Boerman (Radboud University Nijmegen Medical Centre, Netherlands) and Luigi Aloj (Consiglio Nazionale delle Ricerche, Naples, Italy), were cultured in DMEM with GlutaMAX-I, supplemented as above with 250 μg/mL G418 (Geneticin; Gibco BRL Life Technologies) for transfected cells (22). All culture reagents were from Gibco BRL Life Technologies or Biochrom KG Seromed. Cells were maintained in a humidified 5% CO2 atmosphere at 37°C and passaged (1:3 or 1:4) every 2–5 days using 0.05% trypsin–0.02% ethylenediaminetetraacetic acid (w/v) solution.
Metabolic Studies in Mice
Test 111In-labeled radiopeptide solution (100 μL; 11–22 MBq; 3 nmol peptide in physiologic saline–ethanol [9:1, v/v]), with vehicle (100 μL saline–ethanol [9:1, v/v]) (control) or PA (100 μL vehicle with 300 μg PA) (PA-treated), was injected intravenously into tail veins of healthy Swiss albino mice (National Center for Scientific Research “Demokritos” Animal House). Five minutes post-injection, blood (0.5–1 mL) was collected from the heart into pre-chilled polypropylene tubes containing ethylenediaminetetraacetic acid, kept on ice. For [111In-DOTA]MG11, tubes also contained 1 mM methionine to prevent oxidation. Blood samples were centrifuged (10 min, 2,000g, 4°C; Hettich Universal 320R), plasma collected, mixed 1:1 (v/v) with chilled acetonitrile, and centrifuged again (10 min, 15,000g, 4°C). Supernatants were concentrated under N2 at 40°C, diluted with saline (≈400 μL), and filtered (Millex GV 0.22 μm).
Aliquots were analyzed by reversed-phase HPLC using a Symmetry Shield RP18 column (5 μm; 3.9 × 20 mm; Waters). Elution was performed over 50 minutes at 1 mL/min with a gradient from 100% A (0.1% trifluoroacetic acid in H2O) and 0% B (acetonitrile) to 50% A and 50% B. Intact radiotracer elution times were determined by coinjection of the respective 111In-labeled radiopeptide.
Biodistribution of 111In-Labeled Radioligands in Xenograft-Bearing SCID Mice
Six-week-old SCID mice (National Center for Scientific Research “Demokritos” Animal House) were inoculated in flanks with ≈150 μL bolus containing 1 × 107–2 × 107 freshly harvested cells in sterile saline. Animals were kept under aseptic conditions until palpable tumors developed. Tumor development times were 12 days for AR4-2J (350 ± 20 mg [mean ± SD]), 3 weeks for PC-3 (150 ± 30 mg), and 6 days for A431-CCKR(+) or A431-CCKR(−) tumors (260 ± 80 mg).
On the experiment day, test 111In-labeled radiopeptide solution (100 μL; 37–74 kBq; 10 pmol peptide in vehicle), with vehicle (100 μL) (control), PA (100 μL vehicle with 300 μg PA) (PA-treated), or PA plus receptor blocker (100 μL vehicle with 300 μg PA and excess receptor blocker) (receptor blockade), was injected intravenously. For [111In-DOTA]MG11, 1 mM methionine was included. For receptor blockade, animals received PA co-injected with 40 nmol [Tyr3]octreotate (sst2 blockade), 40 nmol [Tyr4]bombesin (GRPR blockade), or 40 nmol demogastrin 2 (CCK2R blockade). Animals were randomly assigned to groups. Two additional AR4-2J tumor-bearing groups received [111In-DTPA]octreotide with vehicle (control) or PA (PA-treated).
Groups of 4 animals were euthanized at 4 hours post-injection. Blood, urine, and organs were collected, weighed, and radioactivity measured using an automatic well-type γ counter. Data were calculated as percentage injected dose per gram of tissue (%ID/g) using standard solutions and Microsoft Excel. Results are mean ± SD, calculated using PRISM 2.01 (GraphPad).
Statistical analysis used an unpaired 2-tailed Student t-test to compare control, PA-treated, and receptor blockade groups at 4 hours. P values < 0.05 were considered significant.
SPECT/CT Imaging of [111In-DOTA]MG11 in AR4-2J Tumor-Bearing Nude Mice
NMRI nu/nu mice (≈30 g; Harlan CPB) were subcutaneously inoculated with 106 AR4-2J cells (200 μL injection volume) in the shoulder region. When tumors reached ≈1 cm diameter, animals were scanned under isoflurane anesthesia with body temperature maintained. Post-examination, animals were euthanized.
All animals received [111In-DOTA]MG11 (45 MBq; 0.5 nmol) with vehicle or PA (300 μg) intravenously 4 hours before SPECT imaging (200 μL total volume). SPECT/CT images were acquired using a 4-head multipinhole SPECT/CT camera with 111In energy peak settings at 171 and 245 keV (± 10% window). Nine-pinhole apertures (1.4 mm diameter) were used per camera head (230 × 215 mm detector). Matrix size was 256 × 256. Animals were centered in rotation; rotation radius was fixed. SPECT images were acquired with 20 projections, 60 s per projection, and a 0.8 quality factor. Reconstruction used ordered-subset expectation maximization with HiSPECT NG software (Scivis Wissenschaftliche Bildverarbeitung GmbH), 24 iterations, and 0.3 × 0.3 × 0.3 mm voxel size. CT images were acquired with 180 projections, 45-kVp tube voltage, 500-ms exposure time, and 0.2 × 0.2 × 0.2 mm voxel size, and reconstructed using exact-cone-beam filtered backprojection. SPECT images were resampled to CT resolution and registered using VivoQuant program (version 1.22; inviCRO LLC).
All animal experiments complied with European and national regulations and were approved by national authorities.
RESULTS
111In-Labeled Radiotracers
111In incorporation into DOTA-conjugated peptides was achieved by heating at 90°C for 20 minutes in acidic media, consistent with prior findings (26). Reversed-phase HPLC analysis confirmed that all three 111In-labeled radiotracers were eluted as single radiochemical species (>95% purity).
Effect of PA on In Vivo Stability and AR4-2J Tumor Uptake of [111In-DOTA-Ala1]SS14 in Mice
Figure 1 illustrates PA co-injection effects on [111In-DOTA-Ala1]SS14 stability and tumor localization in mice. Blood analysis 5 minutes post-injection revealed near-complete radiopeptide degradation (Figure 1A) (16,31). However, PA co-injection increased parent radiopeptide levels in blood to 86% (Figure 1B). Crucially, this enhanced circulating intact radiotracer significantly amplified radiolabel levels in AR4-2J tumors at 4 hours. sst2-expressing tumor uptake increased from below 1 %ID/g to 14 %ID/g (Figure 1C and inset), while uptake decreased to <0.3 %ID/g with PA and excess [Tyr3]octreotate co-injection, indicating sst2-mediated uptake. In contrast, metabolically stable [111In-DTPA]octreotide uptake in AR4-2J tumors (n = 7) at 4 hours was 5.5 ± 1.0 %ID/g in the control group and 5.6 ± 1.0 %ID/g in the PA-treated group, showing no PA effect on stable peptides.
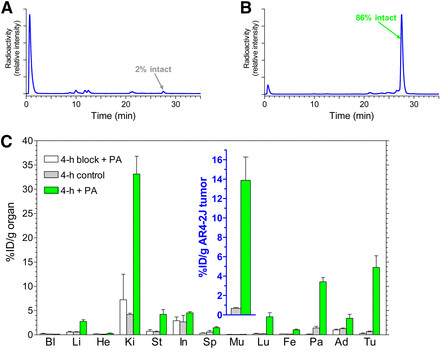 FIGURE 1.
FIGURE 1.
FIGURE 1.(A and B) HPLC analysis of blood collected 5 min after injection of [111In-DOTA-Ala1]SS14 into healthy mice without (A) or with (B) coinjection of PA. (C) Tissue distribution data at 4 h after injection of [111In-DOTA-Ala1]SS14 without PA, with PA, or with PA plus excess sst2 blocker coinjection into male SCID mice bearing AR4-2J xenografts; values for tumors are shown in inset. Ad = adrenal glands; Bl = blood; Fe = femur; He = heart; In = intestines; Ki = kidneys; Li = liver; Lu = lungs; Mu = muscle; Pa = pancreas; Sp = spleen; St = stomach; Tu = AR4-2J tumors.
Effect of PA on In Vivo Stability and Human PC-3 Xenograft Uptake of 111In-PanSB1 in Mice
Figure 2A shows rapid 111In-PanSB1 breakdown within 5 minutes of entering healthy mouse bloodstream. PA co-administration dramatically increased circulating 111In-PanSB1 levels from 12% to 80% (Figure 2B). Consistently, radioactivity uptake in GRPR-positive PC-3 xenografts in SCID mice escalated from <4 %ID/g to >21 %ID/g at 4 hours with PA co-injection (Figure 2C and inset). Uptake in xenografts reduced to <0.9 %ID/g with PA and excess [Tyr4]bombesin co-injection, confirming GRPR specificity.
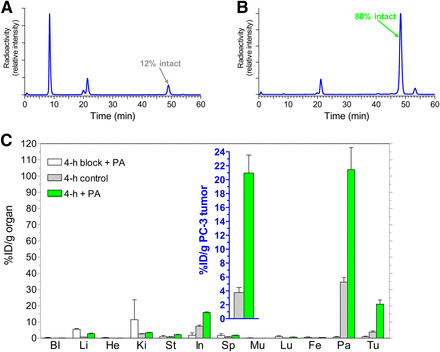 FIGURE 2.
FIGURE 2.
FIGURE 2.(A and B) HPLC analysis of blood collected 5 min after injection of 111In-PanSB1 into healthy mice without (A) or with (B) coinjection of PA. (C) Tissue distribution data at 4 h after injection of 111In-PanSB1 without PA, with PA, or with PA plus excess GRPR blocker coinjection into female SCID mice bearing human PC-3 xenografts; values for tumors are shown in inset. Bl = blood; Fe = femur; He = heart; In = intestines; Ki = kidneys; Li = liver; Lu = lungs; Mu = muscle; Pa = pancreas; Sp = spleen; St = stomach; Tu = PC-3 tumors.
Effect of PA on In Vivo Stability and A431-CCK2R(+) Tumor Uptake of [111In-DOTA]MG11 in Mice
HPLC analysis of blood 5 minutes post-[111In-DOTA]MG11 injection showed only 5% intact radiopeptide in mouse blood (Figure 3A). PA treatment significantly increased circulating [111In-DOTA]MG11 to 70% (Figure 3B). The impact on tumor uptake was studied in a double-tumor model in SCID mice (23). PA co-injection dramatically increased uptake, from 2 %ID/g to >15 %ID/g, specifically in A431-CCK2R(+) tumors, not in A431-CCK2R(−) tumors (Figure 3C and inset). CCK2R-negative tumors showed minimal uptake (<0.5 %ID/g), confirming CCK2R-specific targeting enhancement by PA.
 FIGURE 3.
FIGURE 3.
FIGURE 3.(A and B) HPLC analysis of blood collected 5 min after injection of [111In-DOTA]MG11 into healthy mice without (A) or with (B) coinjection of PA. (C) Tissue distribution data at 4 h after injection of [111In-DOTA]MG11 without PA or with PA coinjection into male SCID mice bearing A431-CCK2R(+) and A431-CCK2R(−) xenografts; values for tumors are shown in inset. Bl = blood; Fe = femur; He = heart; In = intestines; Ki = kidneys; Li = liver; Lu = lungs; Mu = muscle; Pa = pancreas; Sp = spleen; St = stomach; Tu − = A431-CCK2R(−) tumors; Tu + = A431-CCK2R(+) tumors.
Effect of PA on AR4-2J Tumor Uptake of [111In-DOTA]MG11 in Mice: Visualization with SPECT/CT
PA treatment significantly increased [111In-DOTA]MG11 uptake in AR4-2J tumors (expressing rat CCK2R), from <2 %ID/g to >16 %ID/g at 4 hours. With PA and excess demogastrin 2 (24) co-injection, tumor uptake decreased to 0.3 %ID/g, indicating CCK2R-mediated accumulation (Figure 4A and inset). SPECT/CT imaging of AR4-2J tumor-bearing mice corroborated these findings, demonstrating improved tumor visualization due to PA (Figures 4B and 4C).
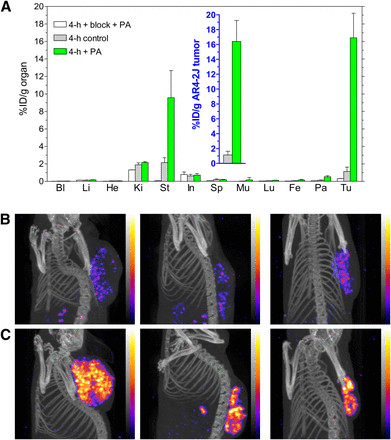 FIGURE 4.
FIGURE 4.
FIGURE 4.(A) Biodistribution of [111In-DOTA]MG11 at 4 h after injection without PA, with PA, or with PA plus excess CCK2R blocker coinjection into AR4-2J tumor–bearing mice; values for tumors are shown in inset. Bl = blood; Fe = femur; He = heart; In = intestines; Ki = kidneys; Li = liver; Lu = lungs; Mu = muscle; Pa = pancreas; Sp = spleen; St = stomach; Tu = AR4-2J tumors. (B and C) SPECT/CT at 4 h after injection of [111In-DOTA]MG11 without (B) or with (C) coinjection of PA (n = 3 per group). Note the highly improved tumor visualization in C.
DISCUSSION
The past two decades have witnessed significant advancements in experimental and clinical oncology. Notable progress includes the detailed characterization of peptide receptor expression on cancer cell surfaces at levels exceeding those in surrounding healthy tissues. Peptide receptors, belonging to the serpentine G protein-coupled receptor (GPCR) superfamily, are increasingly recognized for their roles in cancer (1–3). Nuclear medicine researchers have capitalized on these advancements, developing novel molecular tools based on radiolabeled peptide ligands to localize and treat primary and metastatic cancers. Radiopeptides are designed to carry radionuclides, suitable for SPECT (gamma emitters) or PET (positron emitters) imaging, or for radionuclide therapy (alpha, beta, or Auger electron emitters), to GPCR targets with high specificity (4,6).
The clinical success of radiolabeled somatostatin analogs in diagnosing, staging, and treating neuroendocrine tumors overexpressing sst2 is particularly relevant. This success relies heavily on the metabolic stability of the radiopeptides used. Unlike rapidly degradable native somatostatin-14 (31), sst2-targeted radiopeptide drugs like [111In-DTPA]octreotide are based on robust cyclic octapeptide analogs like octreotide (7–9). While neuroendocrine tumor prevalence is relatively low, other GPCR targets for radiopeptide ligands are abundantly expressed in more common human cancers (3). For example, GRPRs are highly expressed in prostate, breast, and lung cancers and are potential targets for bombesin-like radioligands (32,33). Expanding the clinical impact of radiopeptides hinges on translating the success of [111In-DTPA]octreotide to other radiopeptide families.
However, limited success persists due to the poor metabolic stability of many radiopeptide drugs. Structural modifications aimed at improving stability often compromise receptor affinity or pharmacokinetic profiles (10,11). Furthermore, standard in vitro metabolic stability assays, using mouse or human serum, often overestimate stability by neglecting enzyme activity associated with vasculature, blood cells, and major organs (34).
This study explored a novel approach to enhance tumor targeting: in vivo co-administration of an enzyme inhibitor and a radiopeptide. This “serve to protect” strategy aims to safeguard the radiopeptide from degradation upon entering circulation, ensuring its protected transit to tumor-associated receptors. Transient inhibition is sufficient to protect the radiopeptide during its journey to the target. We hypothesize that increased tumor uptake from this strategy will improve diagnostic sensitivity and therapeutic efficacy of radiopeptide drugs in cancer patients.
We demonstrated the feasibility of this approach using radiopeptides from the somatostatin, bombesin, and gastrin families. Co-injection of phosphoramidon (PA), a single NEP inhibitor (12,13), with test radiopeptides led to a marked increase in tumor uptake in mouse models, directly correlated with improved circulating levels of intact drug. Importantly, PA had no effect on the tumor uptake of metabolically stable [111In-DTPA]octreotide in the same sst2-positive model used for [111In-DOTA-Ala1]SS14. This supports our central hypothesis that enhanced tumor uptake results from improved radiopeptide bioavailability via NEP inhibition by PA. The significant impact of a single NEP inhibitor on radiopeptide bioavailability and tumor targeting is notable, given the vast array of proteases in the human body. The human degradome includes at least 569 proteases across five classes: metalloproteinases, serine proteases, cysteine proteases, threonine proteases, and aspartic proteases. Mouse and rat degradomes are even larger, with 644 and 629 members, respectively (35,36). While NEP appears crucial in initiating the breakdown of tested radiopeptides, and potentially others, we continue to investigate other potential enzymes involved.
This proposed strategy holds significant promise for biodegradable radiopeptides, including those based on native sequences (16,31, which are evolutionarily optimized for receptor interaction. This approach is expected to broaden the application of radiopeptide analogs across more cancer types. Furthermore, it provides a versatile method for identifying enzymes responsible for in vivo degradation of specific radiopeptide drugs. This information is crucial for rational design of stabilized analogs and could also benefit other peptide drugs, such as peptide-drug conjugates, optical imaging probes, or phage display-selected peptides (37).
CONCLUSION
This study underscores the critical importance of radiopeptide in vivo stability for successful tumor targeting in cancer visualization and therapy. Our findings reveal that a single peptidase, NEP, plays a significant role in the rapid in vivo breakdown of intravenously administered radiopeptides from the somatostatin, bombesin, and gastrin families.
Most importantly, our results offer exciting opportunities to “serve to protect” biodegradable radiopeptides, enhancing their delivery and tumor accumulation by simply co-injecting a protease inhibitor like PA. This amplification of radiolabel levels in tumor lesions is expected to significantly boost diagnostic sensitivity and therapeutic efficacy. The extensive existing knowledge of peptidase inhibitors, including some already tested in human trials or approved as drugs, highlights the translational potential of this “serve to protect” strategy for clinical practice.
The novel concept of radiopeptide drug-enzyme inhibitor co-injection to optimize tumor uptake is particularly relevant for molecular imaging and therapy. Nuclear medicine protocols involve single or limited systemic drug administrations, requiring only short-term enzyme inhibition. Protection is primarily needed between intravenous radiopharmaceutical injection and tumor receptor target delivery – a brief period for small, rapidly localizing radiopeptide ligands. Water-soluble agents that “escort” radiopeptides through hydrophilic body compartments and induce fast, transient enzyme inhibition offer an elegant route to optimize tumor targeting.
DISCLOSURE
The publication costs for this article were partially defrayed by page charges. This article is therefore marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact. No potential conflicts of interest relevant to this article were reported.
Acknowledgments
We thank Aikaterini Tatsi, Panteleimon J. Marsouvanidis, and Aikaterini Kaloudi for their contributions to metabolic and biodistribution experiments, and Saskia Berndsen, Stuart Koelewijn, Jan de Swart, and Harald Groen for expert support during the SPECT/CT study.
Footnotes
-
Published online Nov. 28, 2013.
-
© 2014 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
-
Received for publication July 15, 2013.
-
Accepted for publication September 25, 2013.
-
1 Reubi JC, Schaer JC, Waser B. Cholecystokinin receptors in the human brain and peripheral tissues. J Nucl Med. 1997;38(7):93N-100N.
-
2 Lamberts SW, Krenning EP, Reubi JC. The role of peptide receptors in the diagnosis and treatment of cancer. Endocr Rev. 1991;12(4):450-482.
-
3 Reubi JC. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr Rev. 2003;24(4):389-427.
-
4 Breeman WA, de Jong M, Bernard BF, et al. Preclinical evaluation of (177)Lu-DOTA-sst2-antagonist for somatostatin receptor-targeted radionuclide therapy. J Nucl Med. 2011;52(7):1075-1082.
-
5 Wild D, Macke HR, Angelberger P, et al. (90)Y-DOTATOC in patients with neuroendocrine tumours: results of phase II study. Eur J Nucl Med Mol Imaging. 2002;29(2):167-176.
-
6 Kwekkeboom DJ, de Herder WW, Kam BL, et al. Treatment with the radiolabeled somatostatin analog [177Lu-DOTA0,Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26(13):2124-2130.
-
7 Krenning EP, Kwekkeboom DJ, Bakker WH, et al. Somatostatin receptor scintigraphy with indium-111-DTPA-D-Phe-1-octreotide: a method for visualizing neuroendocrine tumors. Eur J Nucl Med. 1993;20(8):716-731.
-
8 Kwekkeboom DJ, Krenning EP. Somatostatin receptor radionuclide therapy in neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21(1):1-22.
-
9 Bushnell DL, O’Dorisio TM, O’Dorisio MS, et al. Phase III trial of 90Y-DOTATOC vs high-dose octreotide cold therapy in patients with advanced midgut neuroendocrine tumors: a preliminary report. J Nucl Med. 2010;51(suppl 2):1814.
-
10 Verber DF, Freidinger RM. Design of metabolically stable peptide analogs. Trends Neurosci. 1985;8(9):351-355.
-
11 Lipton MA, Kaiser ET. Synthetic peptide combinatorial libraries for drug discovery. Biopolymers. 1995;37(2):127-140.
-
12 Turner AJ, Tanzawa K. Mammalian membrane metallo-endopeptidase: NEP and related proteins. FASEB J. 1997;11(5):355-364.
-
13 Beaumont A, Barrett AJ, Miller CJ. The active site of neprilysin (neutral endopeptidase EC 3.4.24.11). Bioessays. 1995;17(1):35-44.
-
14 Miyake R, Shibazaki M, Kanai M. Catalytic enantioselective synthesis of phosphoramidon. J Am Chem Soc. 2004;126(5):1620-1621.
-
15 Wosikowski K, Reichwein JF, Steckler M, et al. Large-scale synthesis of phosphoramidon. Org Process Res Dev. 2007;11(3):542-545.
-
16 Maina T, Nock BA, Nikolopoulos D, et al. [111In-DOTA-DPhe1]octreotide, a promising radiopharmaceutical for tumour imaging and therapy: preclinical evaluation in animal models. Eur J Nucl Med. 2000;27(10):1503-1511.
-
17 Ginj M, Chen J, Froidevaux S, et al. Bombesin receptor subtypes in human prostate cancer: evaluation of больных using bombesin analogues. J Nucl Med. 2001;42(1):78-86.
-
18 Cescato R, Maina T, Maecke HR, et al. Bombesin receptor subtype-2 (BB2R)-selective agonists induce intracellular calcium mobilization and ERK1/2 phosphorylation in BB2R-transfected Chinese hamster ovary cells. J Pharmacol Exp Ther. 2001;299(1):408-415.
-
19 Waser B, Tamma R, Cescato R, et al. Highly efficient internalisation of receptor-bound agonists is essential for potent agonistic activity on bombesin receptor subtype 2. Gut. 2005;54(8):1107-1116.
-
20 Reubi JC, Waser B, Schaer JC, et al. Bombesin receptor subtypes in human cancers: detection with receptor autoradiography. J Nucl Med. 2002;43(5):640-646.
-
21 Markwalder R, Reubi JC. Bombesin-like peptides induce intracellular calcium release in small cell lung carcinoma cells. FEBS Lett. 1991;291(1):117-119.
-
22 Becker M, Béhé MP, Haag P, et al. CCK-B/gastrin receptor expressing tumours: in vivo SPECT with [99mTc]-CCK-8 and in vitro validation. Eur J Nucl Med Mol Imaging. 2003;30(1):33-41.
-
23 Nikolopoulos D, Anagnostopoulos C, Pelecanou M, et al. Development of [111In-DOTA]-demogastrin 2, a new radioligand with high tumour uptake and low kidney accumulation for CCK-B receptor-positive tumour imaging. Eur J Nucl Med Mol Imaging. 2006;33(8):948-956.
-
24 Virgolini I, Angelberger P, Li SR, et al. Indium-111-DTPA-gastrin for visualization of gastrin-releasing peptide receptor expressing tumors: first clinical results. J Nucl Med. 1995;36(9):1487-1493.
-
25 Christoforidis S, Papachristofilou A, Tsigkos S, et al. Development of a truncated gastrin analogue with improved receptor affinity and low kidney uptake for CCK-B receptor-positive tumour imaging. Nucl Med Biol. 2009;36(6):683-691.
-
26 Breeman WA, van Gameren A, de Jong M, et al. Radiolabeling of peptides and proteins with indium-111 via DTPA or DOTA using microwave heating. J Nucl Med. 2000;41(9):1617-1621.
-
27 Verwijnen WF, Breeman WA, van den Bosch RC, et al. Renal toxicity of radiolabelled peptides and protecting effect of lysine: preclinical findings and clinical implications. Eur J Nucl Med Mol Imaging. 2003;30(3):391-399.
-
28 Otte A, Mueller-Brand J, Dellas S, et al. Yttrium-90-DOTA-D-Phe1-Tyr3-octreotide for treatment of inoperable neuroendocrine tumors. Eur J Nucl Med. 1999;26(11):1439-1447.
-
29 Christophe J, Svoboda M, Calderon-Dominguez M, et al. Entero-pancreatic hormone receptors in rat pancreatic acinar cells. I. [3H]Secretin: specific binding and biological effects. Eur J Biochem. 1976;63(1):31-38.
-
30 Logsdon CD, Williams JA. Pancreatic acinar cell receptors for cholecystokinin and other secretagogues. Am J Physiol. 1983;244(5):G675-G680.
-
31 Bauer W, Briner U, Doepfner W, et al. SMS 201-995: a very potent and long acting somatostatin analogue. Life Sci. 1982;31(21):2211-2218.
-
32 Fleischmann A, Waser B, Reubi JC. Bombesin receptor subtypes in human normal and cancerous tissues: preferential expression of subtype 1 in cancers. Int J Cancer. 2004;109(4):539-547.
-
33 Jensen RT, Battey JF, Spindel ER, et al. International Union of Pharmacology. LIII. Bombesin receptors. Pharmacol Rev. 2008;60(3):309-347.
-
34 Laverman P, Boerman OC, Corstens FH, et al. Radiolabeled peptides for tumor imaging and therapy. Eur J Nucl Med Mol Imaging. 2002;29(5):681-691.
-
35 Puente XS, Sánchez-Pulido L, Velasco G, et al. A genome-wide analysis of human and mouse proteases. Nat Rev Genet. 2003;4(7):544-558.
-
36 Overall CM, Blobel CP. In search of partners: linking extracellular proteases to cell surface receptors. Nat Rev Mol Cell Biol. 2007;8(3):245-257.
-
37 Arap W, Pasqualini R, Ruoslahti E. Cancer: targeting the tumor vasculature. Nature. 1998;392(6679):257-270.